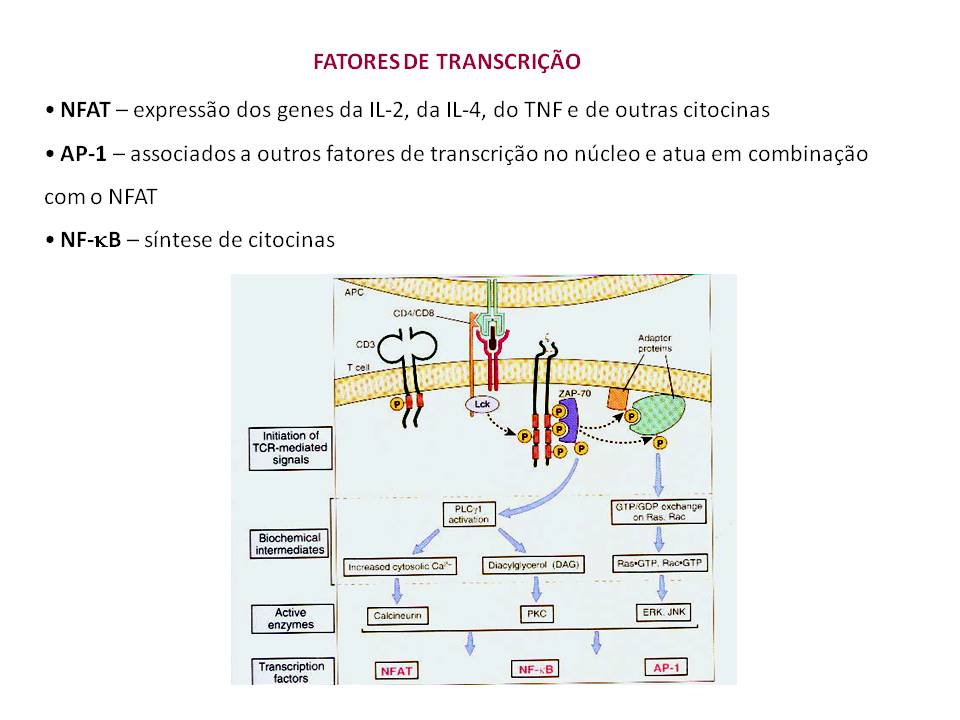
Técnicas laboratoriais de biologia molecular
Soluções Básicas Utilizadas no
Laboratório de Biologia Molecular:
Tris.HCl
[tris(hydroxymethyl)aminomethane] - 1M
Dissolva
121g de Tris Base em 800 ml de água MiliQ
Ajuste o ph desejado
com HCl concentrado
Adicione água até 1L
Autoclave
Cerca de 70 ml de HCl são necessários para pH de 7,4 ; e cerca de 42 ml
para pH de 8,0.
Nota importante: O pH dos
tampões com Tris muda significativamente com a temperatura, caindo cerca de
0,028 U para elevação de cada 1oC. O pH das soluções tamponadas com
Tris deve ser ajustado na temperatura na qual as soluções serão usadas. Como o
pK do Tris é 8,08, o tampão não deve ser usado a pH abaixo 7,2 nem acima de
9,0.
Não adicione
DEPC (Diethyl Pyrocarbonate) em soluções com Tris, porque o Tris inativa essa
substância. Se a solução deve ser usada em experimentos com RNA, prepare-a com
água previamente tratada com DEPC e autoclavada.
EDTA (ethylenediamine tetra-acetic
acid) - 0,5M (pH 8,0)
Dissolva
186,1g de Na2EDTA.2H20 em 700 ml de água
Ajuste o pH para 8,0
com NaOH 10M (cerca de 50 ml)
É difícil dissolver o sal. Ele só entra em solução
quando o pH já está próximo de 8.
Adicione água MiliQ
para 1 L.
Autoclave
TE (tampão Tris/EDTA)
10 mM Tris.HCl - pH 7,4
- 7,5 ou 8,0
1 mM EDTA
SSC (sodium chloride/sodium
citrate), 20X
NaCl 3M - 175g/L
Na3citrate.2H2O 0,3M - 88g/L
Ajuste o pH para 7,0
com HCl 1M
TBE(Tris/Borate/EDTA) - Tampão
de Eletroforese
10X - 1L de solução
stock
108 g de Tris Base (890
mM)
55 g de ácido bórico
(890 mM)
40 ml de EDTA 0,5M pH 8,0 (20 mM)
TAE(Tris/Acetate/EDTA) -
Tampão de Eletroforese
50 X - 1L de solução
stock
242 g de Tris Base
57,1 ml de ácido
acético g;acial
37,2 g Na2EDTA.2H2O
Água MiliQ para 1 L
NaCl - 5M
SDS (Sodium Dodecyl Sulfate) -
10%
Prepare todas as soluções com
água da melhor qualidade possível
Destilada - Deionizada (MiliQ).
FUNDAMENTOS
TEÓRICOS
Extração de
DNA:
Embora o DNA em forma de dupla fita seja muito estável
e resistente a condições adversas, o DNA é vulnerável mesmo a forças leves
originadas durante a pipetagem ou agitação que facilmente geram tensão na
molécula e quebra das ligações fosfodiéster. A obtenção de moléculas de DNA com
mais de 150 kb não é fácil. No entanto, é muito fácil extrair moléculas de DNA
fragmentadas, com 50 a 100 kb.
A quantidade de DNA que se obtém usualmente a partir
de 20 ml de sangue é ~250 mg.
A extração de DNA inclui basicamente os seguintes
procedimentos:
1.
Lise das células (citólise)
2.
Purificação do DNA, separando-o
de macromoléculas contaminantes, tais como proteínas e RNA, por digestão
enzimática e/ou processos físico-químicos.
O local no laboratório e o material utilizados para
extração de DNA devem ser separados daqueles dedicados ao manuseio de DNA
clonado em plasmídeos ou outro tipo de vetor. Contaminação do DNA genômico, no
processo de extração, com esse tipo de DNA poderá causar sérios problemas em
experimentos futuros. Uma possível fonte de contaminação é o uso comum de
centrífugas.
Extração de DNA de sangue:
Anticoagulante:
1 - citrato: (solução ACD)
0,48% (peso/volume) - ácido cítrico
1,32% - citrato de sódio
1,47% - glicose
Use 3,5 ml para cada ~20 ml de sangue.
2 - EDTA - pode ser usado.
A solução ACD é ideal, se o objetivo é preparar DNA genômico a partir de
sangue total, pois preserva melhor moléculas de DNA de elevado peso molecular.
Tanto em citrato como em EDTA, o sangue pode ser estocado por vários
dias a 0oC ou indefinidamente a -70oC, antes da extração
do DNA. O sangue não deve ser coletado em heparina, pois heparina pode inibir a
reação de PCR que eventualmente poderá ser realizada mais tarde.
1 - Tranfira o sangue para um tubo de centrífuga e centrifugue a 1300 g
por 15 minutos a 4 oC.
2 - Remova o
sobrenadante. Transfira, com uma pipeta Pasteur ou ponteira P-1000, a camada
superior de células, correspondente aos leucócitos, para um outro tubo.
Descarte o pellet de hemácias.
3 - Centrifugue novamente a porção correspondente aos leucócitos. Remova
o sobrenadante residual. Ressuspenda o pellet em 15 ml de tampão de lise.
Incube a solução a 37oC por 1 hora.
Solução de lise:
Tris-HCl,
pH 8,0 - 10 mM
EDTA,
pH 8,0 - 10 mM
SDS
- 0,5%
RNase-DNase free - 20 mg/ml
A solução sem a
RNase é preparada e deixada em temperatura ambiente. RNase é adicionada em uma
alíquota só no momento do uso.
Se for sangue
congelado:
1 - Descongele o
sangue em um banho maria a temperatura ambiente. Adicione igual volume de uma
solução salina tamponada com fosfato (PBS), à temperatura ambiente.
2 - Transfira a
mistura para um tubo para centrifuação. Centrifugue a 3.500g, po 15 minutos, a
4 oC.
3 - Remova o
sobrenadante que contém as hemácias lisadas. Ressuspenda o pellet em 15 ml de
tampão de lise.
Incube a solução a 37oC por 1 hora.
Na fase
de lise é essencial que toda a amostra entre em contato com o tampão de lise.
Este deve estar sempre em quantidade suficiente e a homogeneização deve ser tal
que garanta realmente o contato de todas as células com o tampão de lise.
4 - Transfira o material lisado para outros tubos de centrifugação, de
tal modo que ocupe apenas 1/3 do tubo.
Adicione proteinase K (20 mg/ml) a uma concentração final de 100 mg/ml. Misture delicadamente.
Pode-se usar um bastão de vidro para isso.
Certifique-se de que a proteinase K é de boa
procedência (genomic grade – livre de DNases e RNases), está dentro do tempo de
validade e não foi estocada de forma de incorreta. O melhor é preparar a
solução estoque de proteinase K (20 mg/ml) a partir do liofilizado quando for
usar. Se for estocar, separe em alíquotas e guarde a –20oC.
5 - Incube em um banho maria a 50oC por 3 horas. Misture
algumas vezes durante este período.
6 - Resfrie a solução até a temperatura ambiente e adicione igual volume
de fenol equilibrado com Tris-HCL - 0,1 M (pH 8.0). Misture delicadamente as
duas fases - para isso ponha o tubo com boca para baixo em um agitador de tubos
por cerca de 10 minutos (agitação leve - não vortex). Alternativamente, inverta
o tubo várias vezes, delicadamente.
É importante que o pH do fenol seja ~ 8,0. Se for mais ácido, o DNA
poderá ficar na interface.
A qualidade do fenol é outro ponto crítico. Após
equilibrado com Tris o fenol não pode ser estocado por anos. Ele tende a ser
oxidado, e neste estado levará a quebras na cadeia de DNA, além disso o
rendimento da extração será muito ruim.
7 - Separe as fases por centrifugação a 5.000 g, 15 minutos, temp.
ambiente.
8 - Com uma pipeta graduada larga, transfira o sobrenadante (que é uma
solução viscosa) para um outro tubo. Faça isso delicadamente, sem perturbar a
interface e sem submeter o DNA a choques mecânicos.
9 - Repita a extração com fenol por mais duas vezes.
Se for necessário obter moléculas na faixa de 150-200 kb, purifique o
DNA por diálise:
- Transfira a fase aquosa do procedimento anterior para um saco de
diálise. Feche o saco, deixando espaço para que o volume possa aumentar 1,5 a 2
vezes durante a diálise.
-
Dialise a solução a 4 oC
em 4 L de tampão de diálise. Troque o tampão 3 vezes, a
cada 6 horas. Para diálise completa é necessário cerca de 24 horas.
Tampão de diálise:
Tris-HCl (pH 8,0) - 50 mM
EDTA (pH 8,0) - 10 mM
Se não for necessário DNA com tamanhos tão elevados, precipite o DNA com
etanol.
10 - Adicione à fase aquosa do passo anterior (extração com fenol) 0,2
volume de acetato de amônium 10 M. Adicione 2 volumes de etanol (100%), a
temperatura ambiente. Misture bem, porém delicadamente.
11 - Imediatamente se forma um precipitado de DNA. Usando uma pipeta
Pasteur com ponta selada em forma de alça, remova o DNA e transfira para outro
tubo.
Se o DNA já estiver muito fragmentado e não for possível pescá-lo desta
maneira, centrifugue a 5.000 g, por 5 minutos, em temperatura ambiente.
Despreze o sobrenadante após a centrifugação.
12 - Lave o DNA precipitado com etanol 70%. Centrifugue novamente e
remova o máximo possível sobrenadante.
Deixe o tubo aberto até que se evapore todo o etanol (alguns minutos).
Não deixe o pellet secar, pois será difícil dissolvê-lo.
13 - Adicione 1 ml de TE (pH 8.0) para cada 0,1 ml de células iniciais.
Deixe agitando levemente a 4 oC, até que o pellet se dissolva
completamente.
Quantifique e verifique a qualidade do DNA em gel de agarose 0,6 %.
Existem numerosos protocolos para extração de DNA, das mais diversas
fontes.
O kits comerciais, com utilização de resinas, são muito convenientes e
fornecem DNA de boa qualidade.
A purificação de ácidos nucleicos com fenol e clorofórmio é um
procedimento rotineiro em laboratórios de biologia molecular. Descreveremos o
procedimento a seguir.
Técnica de extração com
fenol-clorofórmio-alcool isoamílico
Fundamentos
O método mais comumente utilizado para
desproteinização do DNA é a extração com fenol, que desnatura as proteínas de
modo eficiente e, provavelmente, as solubiliza. O clorofórmio também é um bom
desnaturante de proteínas com propriedades um pouco diferentes, que determinam
maior estabilidade da interface entre o solvente e a fase aquosa; a interface
fenol puro-água é bastante instável. A
mistura fenol-clorofórmio reduz o volume da solução aquosa retida na fase
orgânica (comparada ao fenol puro), aumentando o rendimento final. O álcool
isoamílico previne a formação de espuma quando a mistura é agitada em vórtex, o
que torna mais fácil a separação das fases orgânica e aquosa. A proteína
desnaturada forma uma camada na interface das duas fases, separando-se do DNA
que se mantém na fase aquosa.
Em seguida o DNA é precipitado por etanol absoluto, na presença de
concentrações relativamente altas (100 a 500 mM) de um sal de cátion
monovalente. Etanol depleta a camada de hidratação
dos ácidos nucléicos e expõe as cargas negativas dos grupos fosfato.
Contra-íons, como Na+ se ligam, aos grupos carregados reduzindo a
força repulsiva entre as cadeias polinucleotídicas, até o ponto em que possa se
formar um precipitado. O precipitado se formará apenas se houver cátions
monovalentes em quantidade suficiente para neutralizar das cargas dos grupos
fosfato.
A precipitação
com etanol absoluto é necessária para concentrar o DNA e para remover resíduos
de fenol e clorofórmio da solução aquosa desproteinizada. Como a maioria dos
solutos inorgânicos e moléculas orgânicas pequenas são solúveis em etanol 70%,
a lavagem do precipitado com etanol-70% irá dessalinizar o DNA de forma
eficiente. Embora tanto cloreto de sódio, como acetato de sódio e acetato de
amônio sejam eficazes para precipitação do DNA, é mais difícil remover NaCl,
devido a sua menor solubilidade em etanol.
O processo, ao final, fornece uma solução de DNA praticamente livre de
eletrólitos (sais) o que será necessário quando o DNA for utilizado na presença
de tampões cuja composição, bem definida,
é fundamental para o sucesso do procedimento.
Finalmente o DNA, precipitado pelo etanol, é
dissolvido em água.
Roteiro para a extração com
fenol-clorofórmio-álcool isoamílico
Todo o processo deverá ser
executado em tubos de polipropileno (leitosos). Os de poliestireno
(transparentes) não resistem ao fenol-clorofómio.
a) Adicione à solução de DNA, resultante dos processos de extração, um
volume igual da mistura fenol-clorofórmio-álcool isoamílico (10:10:1). Misture bem, porém gentilmente, e centrifugue
por 2 a 5 min (o tempo dependerá do volume: volumes menores, tempos mais
curtos).
b) Remova com cuidado a fase aquosa, que fica por
cima, e a transfira para outro tubo. Se houver precipitado branco na interface
repita a extração com fenol-clorofórmio-isoamil, exatamente como no passo
anterior.
c) Adicione à fase aquosa, separada no passo anterior,
um volume 10 vezes menor de solução de acetato sódio 3M, pH 5,2, ou volume 10
vezes menor de NaCl 5M, ou ainda igual
volume de acetato de amônia 4M. O acetato de amônia favorece a precipitação de
fragmentos maiores de DNA, e deve ser usado quando se quer eliminar da amostra
de DNA pequenos oligonucleotídeos (< 30 pb) que possam estar presentes.
c) Ao volume obtido após a adição do acetato de sódio
adicione agora 2 a 2,5 volumes de etanol absoluto gelado . Misture e incube em gelo
seco por 5 min ou a -70oC
por 15 minutos ou a -20oC por 30 min (esses são os tempos mínimos, sem limite para o máximo).
d) Centrifugue por 5 minutos em centrífuga com rotor
de ângulo fixo, em alta velocidade
rotação (10.000 a 14.000 rpm); remova o
sobrenadante.
e) Adicione 1 ml de etanol 70%. Misture invertendo a
tubo várias vezes e centrifugue como no passo anterior. Se o DNA a ser
precipitado for pequeno (menos de 200 pares de base) faça a lavagem do “pellet”
com etanol 95%.
f) Remova o sobrenadante e seque o “pellet” de DNA
g) Dissolva o “pellet” de DNA em um volume adequado de
água (a concentração deverá ser menor do que 1 mg/ml) para uso em outras
manipulações enzimáticas; dissolva em TE, pH 8,0, para estocagem por por tempo
indeterminado.
Desproteinização por pérolas
de vidro ou por sílica-gel
. A desproteinização do
DNA pode ser feita também com a utilização de pérolas de vidro (“glass beads”)
ou partículas de sílica-gel ativadas (5ml
para até 5mg de DNA), que ligam o DNA na presença de iodeto de sódio (NaI, 6M, 3 volumes para 1 volume de solução de
DNA). O DNA ligado às pérolas de vidro
ou às partículas de sílica-gel é lavado com
solução contendo Tris.Cl 20 mM, EDTA 1 mM, NaCl 100 mM, pH 7,4, três a quatro vezes para remover proteínas, e
em seguida é eluído com água ou TE pH 8.0 (0,5 mg/ml).
Extração de
RNA
Fundamentos
A maior
dificuldade no processo de extração de RNA é evitar a ação das ribonucleases
(RNases). Estas enzimas são muito estáveis e não requerem cofatores para suas
atividades. O primeiro passo para extração de RNA, portanto, envolve a lise das
células em um ambiente químico que resulte em desnaturação de ribonucleases. Em
seguida o RNA é separado de outras macromoléculas constituintes das células
Além de impedir
a ação de ribonucleases provenientes das células das quais estamos extraindo o
RNA, tem-se de evitar a introdução acidental de traços de RNAses, de outras
fontes. O uso de luvas é imprescindível, pois a pele frequentemente contém
bactérias e fungos que podem contaminar a preparação de RNA e serem fontes de
RNAses. Evitar a contaminação com microorganismos é obrigatório quando se
trabalha com RNA.
Todo material
de plástico utilizado tem de ser esterilizado, livre de RNases e não deve ser
reutilizado. O material de vidro deve ser muito bem lavado, autoclavado e
deixado em estufa a 180oC por pelo menos 8 horas. Alternativamente,
os recipientes a serem utilizados podem ser preenchidos com água contendo
dietil-pirocarbonato (DEPC) a 0,1%, que é um potente inibidor de RNAses. Após serem preenchidos com solução de DEPC,
esses recipientes devem ser incubados a 37°C
por pelo menos 2 horas, lavados várias vezes com água estéril (água-DEPC
autoclavada) e aquecidos a 100°C por 15 minutos ou autoclavados. Com
estes procedimentos o DEPC é removido por evaporação. Traços de DEPC indevidamente persistentes
nos frascos podem modificar resíduos de purinas no RNA por carboximetilação, e,
assim, inteferir na tradução do RNA “in
vitro”. Traços de DEPC também podem inibir algumas enzimas.
As cubas para
eletroforese do RNA devem receber o seguinte tratamento:
n
lavagem com solução contendo
detergente
n
enxague com água (Mili-Q)
n
secagem com etanol (96%)
n
preenchimento com solução de
água oxigenada (H2O2) 3%. O contacto com água oxigenada
deve se estender por 10 min, em temperatura ambiente.
n
enxague repetido bem com água
tratada com DEPC (0,1%) e autoclavagem.
Os pipetadores
devem ser reservados para uso exclusivo em preparações de RNA. Se a
exclusividade não for possível, os pipetadores devem ser desmontados e tratados
com um dos procedimentos acima para remoção de RNAses.
As ponteiras e tubos
Eppendorfs devem ser autoclavados e sem
contaminação por RNAases; não devem ser reutilizados.
A água e todas as soluções usadas para preparação de
RNA devem ser tratadas previamente com DEPC (0,1%) e autoclavadas. Soluções
contendo Tris não devem ser tratadas com DEPC, porque Tris reage com DEPC,
inativando-o. No entanto, as soluções com Tris devem ser preparadas com água
previamente tratada com DEPC e autoclavada.
A criteriosa observância dos procedimentos que evitam
a contaminação com RNAase e microorganismos garante a obtenção de RNA íntegro e
de boa qualidade ao final do processo de extração.
Técnicas para extração de RNA
I. Extração de RNA de células
em cultura, leucócitos ou células de lavado bucal:
A técnica utiliza um detergente suave para promover a
lise das células. O roteiro é o que se segue
1. Lavar as células com PBS gelado (por 3 vezes) para remover meio de
cultura ou outra solução em que as células estiveram previamente. Para formar o
“pellet” de células, centrifugue a 300g
por 5 min. Partindo de células em
cultura deve se ter um número de células em torno de 2 x 107.
2. Ressuspender as células em 375 ml de
solução de lise gelada (composição dada abaixo). Após agitação cuidadosa, mas vigorosa, a
suspensão deve ficar transparente, indicando citólise.
Composição da solução para lise:
Tris.Cl 10 mM
NaCl
100 mM
MgCl2 5 mM
0,5%
(vol/vol) Nonidet P-40
pH 8
(Prepare com água-DEPC
autoclavada e filtre para esterilizar)
Se as células forem particularmente ricas em RNAses,
use um inibidor de RNases, ou o inibidor de RNase de origem placentária
(RNAsin), na concentração de 1000 U/ml, ou o complexo vanadyl-ribonucleosideo,
na concentração de 10 mM.
3. Transferir as células lisadas para um tubo de microcentrífuga (1,5
ml) e centrifugue por 2 min a 4oC (14.000 rpm).
4. Transferir o sobrenadante para um outro tubo de microcentrífuga
contendo 4 ml de SDS 20%. Misture em um vótex.
(O fluido
sobrenadante é o extrato citoplasmático. É levemente turvo, de cor tendendo a
um branco amarelado. O “pellet”, que contém os núcleos e alguns “debris”
celulares, deve ser branco e bem menor que o “pellet” de células inteiras)
5. Adicionar 2,5 ml de proteinase K (20mg/ml). Incubar por 15 minutos a 37oC.
6. Adicionar 400 ml de fenol-clorofórmio-álcool
isoamílico (5:1:0,04). Agitar em vórtex vigorosamente. Centrifugar por 5 min ou mais a 4oC
(14.000 rpm)
(Se houver a
formação de um precipitado muito espesso na interface, com escassa fase aquosa
sobrenadante, centrifugar por mais alguns minutos; se ainda assim não for
possível separar a fase aquosa, remover a fase orgânica (de baixo) e adicionar
mais 400 ml de clorofórmio-álcool isoamílico (sem fenol).
Agitar em vórtex e centrifugar por 2 minutos. Transferir a fase aquosa
sobrenadante para outro tubo).
7. Repetir um vez o procedimento de extração com
fenol-clorofórmio-álcool-isoamílico.
8. Fazer extração com 400ml de clorofórmio-álcool
isoamílico. Transferir a fase aquosa
sobrenadante para outro tubo.
9. Adicionar à fase aquosa obtida (cerca de 400 ml) ,
40 ml (1/10 do volume de material) da
solução de acetato de sódio 3M, pH 5,2.
Acrescentar 1 ml de etanol absoluto e misturar por inversão do frasco.
10. Incubar em gelo por 30 minutos ou deixar no freezer, a -20oC
durante a noite.
11. Recuperar o RNA centrifugando por 15 min a 4oC a 14.000
rpm.
12. Lavar o “pellet” com 1 ml de etanol 75%
13. Secar o “pellet” e ressuspendê-lo em um volume de 50 a 100 ml de água-DEPC autoclavada.
14. Quantificar por espectrofotometria (A260).
15. Guardar o RNA em solução em freezer a -70oC .
2. Extração de RNA de células
em cultura ou em tecidos
Nesta técnica as células ou tecidos são homogeinizados
em meio com tiocianato de guanidina, um dos mais potentes desnaturantes de
proteínas. O método se baseia na característica
de RNA de permanecer na solução aquosa, contendo guanidine thyocianate
4M, pH 4, na presença da fase orgânica
fenol/clorofórmio. Neste pH baixo, a maioria das proteínas e pequenos
fragmentos de DNA (5 a 10 kpb) permanecerão na fase orgânica, enquanto que os
fragmentos maiores de DNA e algumas proteínas ficarão na interface. A
fragmentação do DNA durante o processo de homogeinização (em geral feito com
Polytron) ajuda a remover o DNA da fase aquosa.
Após a homogeinização em tiocianato de guanidina, o
RNA pode também ser separado de DNA e das proteínas por centrifugação em
gradiente de cloreto de césio, explorando o fato de o RNA ser mais pesado que
DNA e proteínas. É uma técnica simples, com pouca manipulação da amostra, mas
requer ultracentrifugação.
Técnica
Solução de
tiocianato de guanidina (GIT) 4M:
GIT para 100 m de solução: 47,26g
Citrato de Sódio 1M, pH 7.0 - 2,5 ml
Sarkosyl (N-laurylsarcosine) ) - 0,5%
Completar para 100 ml com água-DEPC autoclavada
No momento do uso adicionar 8 ml/ml de BetaMercapto Etanol
(BME - Sigma)
1. Adicionar 1 ml de GIT para cada 100g de tecido ou 107
células (cultura de células) e homogeinizar em Polytron (16.000 rpm/15 sec).
2. Adicionar 0,1 ml de acetato de sódio (2M, pH 4,0) para cada 1 ml de
GIT adicionado anteriormente. Misturar muito bem. Adicionar 1 ml de fenol
saturado com água (pH 4,0) para cada 1 ml de GIT, e misturar
vigorosamente. Adicionar 0,2 ml (para
cada 1 ml de GIT) de clorofórmio-álcool isoamílico (24:1). Misturar muito bem em vórtex e incubar essa suspensão
por 15 minutos em temperatura de 0 a 4 oC.
3. Centrifugar por 20 minutos a 4 oC, 10.000 rpm
(alternativamente, 5.000 rpm por 45 minutos) . Transferir a fase aquosa sobrenadante (que deve ter o mesmo volume de
GIT adicionado no início) para um outro
tubo.
4. Precipitar o RNA adicionando um volume igual de isopropanol 100%.
Incubar a amostra por 20 min a -20oC. Centrifugar por 10 min a 4 oC,
a 10.000 rpm. Desprezar o sobrenadante.
5. Dissolver o “pellet” de RNA em 0,3 ml de solução desnaturante (GIT) e
transferir para um tubo Eppendorf de 1,5 ml (se já não estiver em um
Eppendorf).
6. Precipitar o RNA com igual volume (0,3 ml) de isopropanol 100% (20
min a -20oC). Centrifugar novamente por 10 min a 4 oC,
10.000 rpm. Descarte o sobrenadante e seque o “pellet”.
7. Dissolver o RNA em 50-100 ml de
H2O-DEPC autoclavada. Quantificar e armazenar a
-70°C.
Para verificar a pureza do RNA
determinear a razão de absorbância
260/280 (260 nm para ácicos nucleicos e 280nm para proteínas), que deve ser
1,8-1,9.
Ao secar o pellet não permitir que
esse seque completamente, o que dificultará muito a dissolução do RNA. Se secar
exageradamente, as proteínas ainda presentes como contaminantes se dissolverão
com maior facilidade que o RNA, o que resultará em baixas razões 260/280.
A água utilizada para
diluir o RNA para espectrofotometria deve ter pH > 7,5. pH ácido afeta o
espectro de absorção UV do RNA e diminui significativamente a razão 260/280.
Ajuste o pH da água para valores levemente alcalinos adicionando Na2HPO4
para uma concentração final de 1 mM.
Eletroforese
em gel de agarose:
Sob influência de uma diferença de potencial elétrico
átomos, moléculas e partículas com carga, em solução aquosa, migram na direção
do eletródio com carga oposta. Como as moléculas têm cargas e massas variáveis,
a migração em um meio de viscosidade definida se dará em diferentes
velocidades, com separação das várias frações de uma mistura.
A mobilidade eletroforética, que é a velocidade de
migração por unidade de campo elétrico, é um parâmetro característico da
molécula em um determinado meio de eletroforese. Este parâmetro depende da densidade de cargas
da molécula e do seu tamanho. Depende, portanto, do pK do grupo ionizável e do
pH do tampão que disssolve a amostra. Como a viscosidade do gel determina o
atrito, limitando a velocidade de propagação, a mobilidade eletroforética será
dependente da temperatura. O que normalmente analisamos é a mobilidade
eletroforética relativa da molécula, comparando-se as distâncias de migração
com as de uma substância padrão, aplicada na mesma corrida eletroforética. Estas substâncias constituirão marcadores que
tornam a avaliação da separação eletroforética relativamente independente de
variações na diferença de potencial elétrico e do tempo de eletroforese.
As amostras para
eletroforese não poderão conter partículas grandes ou gotículas de óleo em
suspensão, pois estas interferem com a separação por bloquear os poros da
matriz de eletroforese. Pode-se centrifugar as soluções antes de colocá-las no
gel.
Separar substâncias que
têm ou cargas positivas ou cargas negativas, como os ácidos nucleicos, que só
têm cargas negativas, não constitui, em geral, problema. As moléculas como as
proteínas cuja carga resultante dependente do pH do meio terão as migrações
eletroforéticas fortemente influenciadas pelo pH do tampão.
A força iônica
(concentração total de íons) do tampão utilizado deve ser tão baixa quanto
possível (o mínimo que garanta um pH constante), reduzindo-se, desta forma, as
correntes iônicas que fluem pelo gel e a dissipação térmica excessiva (lembrar
que os íons que constituem o tampão também migram eletroforeticamente,
contribuindo para a corrente total).
Para se avaliar o
progresso da separação e reconhecer o fim da corrida, corantes com alta
mobilidade eletroforética são adicionados juntamente com a amostra.
A matriz do gel de
eletroforese deve ter poros de tamanhos regulares e ajustáveis, deve ser
quimicamente inerte. A eletro-osmose, que é o arraste do solvente pela migração
eletroforética do soluto, quando os poros são pequenos, não deve ocorrer, pois
alteraria a mobilidade dos íons.
Os géis de agarose, um polissarídeo extraído de algas
marinhas, são adequados para separar moléculas com diâmetro igual ou superior a
10 nm. Com a remoção da agaropectina, se obtém tipos de agarose com diferentes
graus de pureza e de eletrosmose. As diferentes
agaroses se caracterizam pelo seu ponto de fusão (melting point), que
pode variar entre 35oC e 95oC e pelo grau de eletrosmose
(mr), que depende do número de grupos polares que persistem após a
purificação.
O tamanho dos poros depende da concentração de
agarose. A concentração em geral é dada como peso de agarose por volume de
água. Em geral, géis a 1% têm poros em torno de 150 nm e géis a 0,16% têm poros
em torno de 500 nm. A agarose é dissolvida em água fervendo e forma um gel
quando esfria. Durante esse processo há formação de duplas hélices que se
juntam lateralmente, de modo a formar filamentos finos. Durante a geleificação
há formação de polímeros.
Eletroforese em gel de agarose é o método de escolha
para separação, purificação e identificação DNA e RNA. São utilizados, para
isso, géis horizontais, submarinos: o gel fica diretamente mergulhado no
tampão. O gel de agarose tem menor resolução que o de acrilamida (que veremos
posteriormente), mas tem uma faixa de separação maior (entre 200 pb e cerca de
50 kpb).
Coloração de ácidos nucléicos no gel de agarose:
O DNA ou o RNA podem ser facilmente
visualizados pela coloração com brometo de etídeo em baixas concentrações. O
brometo de etídeo é um corante flourescente, excitado por luz ultravioleta, que
se intercala entre as fitas da dupla hélice de DNA ou de segmentos
antiparalelos complementares dentro de uma mesma molécula de RNA (que é fita
simples). O DNA ou o RNA podem ser
revelados colocando-se o gel no transiluminador com luz ultravioleta (254 nm).
De 1 a 10 ng de DNA ou RNA são necessários para um sinal que se possa perceber
a vista desarmada.
Justamente
por se intercalar entre fitas da dupla hélice, o brometo de etídeo é um agente
mutagênico e deve ser sempre manuseado com luvas, evitando-se qualquer tipo de
contaminação do pesquisador. O lixo contendo brometo de etídeo também necessita
tratamento especial, que comentaremos posteriormente.
Uma alternativa interessante ao brometo de etídeo é o
corante SYBR Green I (para DNA) e SYBR Green II (para RNA) - da Molecular
Probes, que pode ser utilizado tanto em gel de agarose como de poliacrilamida.
Este corante pode detectar até 20 pg de DNA-dupla fita e até 100 pg de RNA ou
de DNA-fita simples. A excitação máxima é obtida a 497nm; em 254 nm a sensibilidade é cerca de 3 a 5 vezes
menor. A emissão tem o pico em 520 nm. O que torna o SYBR green superior ao
brometo de etídeo é a sensibilidade de 50 a 100 vezes maior, podendo detectar
1-2 ng de oligonucleotídeos em gel de poliacrilamida 5% (UV/450 nm). O SYBR
Green tem excepcional afinidade por DNA e apresenta um grande aumento de
fluorescência após a ligação. É muito
menos mutagênico que o brometo de etídeo, mas como ainda são poucos os dados
referentes a sua mutagenicidade.
Técnica
para eletroforese de DNA
Tampões:
Podem ser usados o TBE (Tris/base-Borato-EDTA)
ou TAE (Tri/Acetato-EDTA). Utizamos na
rotina de nosso laboratório o TBE.
Ao preparar as soluções de tampão use
água destilada e deionizada, proveniente de um sistema de purificação tipo Nono-Pure ou MiliQ.
Solução
estoque de TBE concentrada em 10 vezes (por litro);.:
108g de Tris base/l (890 mM)
55g de ácido bórico/l (890 mM)
40 ml de EDTA 0,5 M - pH 8,0 (20 mM)
Concentração de trabalho: diluir
10. Para gel de agarose pode-se usar
diluições de 20 vezes, sem problemas. A
concentração elevada da solução estoque tende a produzir precipitação dos
sais. Pode-se, para evitá-la, preparar
soluções estoque 5 vezes concentrado.
Solução
estoque de TAE concentrada 50 vezes
(para 1 l):
242g de Tris base
57,1 ml de ácido cético glacial (Tris.acetato 2M)
37,2g de Na2EDTA.2H20 (100 mM)
H2O para 1 l
Concentração de trabalho:
diluir 50 vezes (40 mM Tris.acetato; 2 mM Na2EDTA.2H2O;
pH ~8,5)
TAE tem menor poder tamponante, e a capcidade de
tamponamento pode se esgotar se o tempo de eletroforese for muito longo.
Preparação do gel de agarose a 1 %
1. Adicione 100 ml de TBE ou TAE a 1g de DNA Typing Grade Agarose.
2. Misture e aqueça no forno
de micro-ondas por tempo de 1 min a 1 min e 30 seg.
3. Misture, agitando levemente o frasco, e verifique
se não há fragmentos íntegros de agarose. Se houver aqueça por mais alguns
segundos.
4. Coloque a agarose fundida num cilindro graduado de
100 ml e complete o volume com água (alguma água terá evaporado)
5. Retorne a agarose para o frasco anterior e adicione
5 ml de solução de Brometo de Etídeo (10mg/ml). Se for corar com SYBER
Green, não execute este passo.
6. Coloque a agarose na bandeja de eletroforese
previamente vedada e com o pente que formará as pocinhas onde serão colocadas
as amostras de DNA.
7 . Deixe esfriar.
Prepare 1 l de TBE ou TAE para cobrir o gel de agarose na cuba de
eletroforese.
Preparo da amostra de DNA:
1. Separe o volume da amostra de DNA a ser colocado no
gel de eletroforese (de acordo com a quantificação feita previamente)
2. Acrescente água (MilliQ-autoclavada) até o volume
final desejado (por ex.: 18 ml)
3. Acrescente o “loading buffer” concentrado 10 vezes,
de modo a diluí-lo para concentração de trabalho (no exemplo: 2 ul).
“Loading
Buffer”:
Para que a amostra de DNA permaneça nas poças do gel de agarose
(visíveis quando se retira o pente que serve de molde), é necessário que se
acrescente a ela algo que a torne mais densa que o tampão de corrida (ex: Ficoll 400, glicerol ou sacarose). Além disso,
acrescentamos um ou dois corantes de elevada mobilidade eletroforética, que nos
fornecerá informação sobre o andamento da eletroforese, indicando o momento de
interrompê-la.
Protocolos
1- 5ml de loading buffer concentrado 10 vezes:
1 ml de EDTA 0,5 M - pH 8,0
(100 mM EDTA)
2,5 ml de glicerol (50%)
0,025g de azul de bromofenol
(0,5%)
ou
2- loading buffer com TAE - concentrado 2 vezes:
2X TAE buffer pH 8,3
20 Mm EDTA
5% Ficoll 400
0,1% azul de bromofenol
Coloque a amostra no gel, sempre ao
lado de um marcador de peso molecular que deve ser preparado do mesmo modo que
sua amostra. Desligue a eletroforese quando o azul de bromofenol tiver
percorrido cerca de 2/3 do gel ou mais.
Se tiver colocado brometo de etídeo
no gel, visualise o DNA no transiluminador UV e fotografe (Polaroid, filme 667
branco e preto). Utilize uma régua transparente a UV durante a fotografia, para
marcar a posição dos marcadores de peso molecular após transferência do DNA
para a membrana, no passo posterior (“Southern blot”).
Se utilizar SYBR Green, dilua-o a
1:10.000 em TE (10 mM Tris.HCl, 1 mM EDTA, pH 8,0), em TBE ou emTAE, pH entre 7,5 e 8,0 (preferencialmente
8,0). Cubra o gel com o corante diluído
por 10-40 min, dependendo da espessura do gel. Remova o gel do corante,
visualise o DNA no transiluminador UV (254 nm) e fotografe, ou faça o
“scanning” do gel no STORM (Molecular
Dynamic), se dispuser desse equipamento.
Quando se pretende fazer uma análise
posterior dos fragmentos de DNA, separados por eletroforese, é necessário
transferí-los para uma membrana (nylon) para posterior hibridização com sonda
específica (ver protocolo de “Southern blot” mais adiante).
Técnica
para eletroforese de RNA:
Fundamentos
Para a eletroforese de RNA o
tampão usualmente utilizado é o MOPS (3-(N-morpholino)-propanesulfonic acid),
pH 7,0 (o RNA é facilmente hidrolizado em meio alcalino). Como o objetivo mais
freqüente é transferir o RNA para uma
membrana (“northern-blot”), para posterior análise com sondas específicas, a
eletroforese é efeita em condições desnaturantes, porque embora a molécula de
RNA tenha uma única fita, dupla hélice de segmentos antiparalelos
complementares são freqüentes e sua persistência irá dificultar a ligação do
RNA à membrana, assim como a hibridização da sonda com a molécula de RNA que
está sendo analisada. O agente desnaturante mais freqüentemente utilizado é o
formaldeído (embora existam outras opções, como o glioxal)
Técnica
Solução
estoque de MOPS
(10X):
0,4M MOPS, pH 7,0
0,1M
acetato de sódio
0,1M EDTA
Preparação
da solução
A 400 ml de
H20-DEPC (autoclavada) adicione
20,9g de MOPS
8,3 ml de
acetato de sódio 3M - pH 5,2 (solução tratada com DEPC e autoclavada)
Ajuste o pH para 7,0 com “pellets” de NaOH
Eleve o volume final para 500ml
Esterilize
por filtração ou autoclave (se autoclavar, a solução ficará amarelada, o que é
normal).
Armazene a solução protegida da luz (envolva o frasco em papel alumínio)
Gel de agarose:
1. 1,0 a 1,5 g de agarose
2. 10 ml de 10X MOPS
3. 87 ml de H2O-DEPC
autoclavada
4. Dissolva a agarose,
aquecendo em forno de micro-ondas
5. Deixe esfriar em
banho-maria a 50oC
6. Adicione 5,1 ml de
formaldeído a 37% (em capela para produtos químicos tóxicos)
7. Homogeinize muito bem
evitando formar bolhas
8. Coloque na bandeija de
eletroforese, já com o pente, e deixe esfriar.
(A cuba de eletroforese, a bandeja o pente devem ser previamente
tratados para eliminar quaquer contaminação com RNase, como previamente
descrito).
“Loading buffer” para
RNA (1 ml)
1. 0,75 ml
de formamida pura (deionizada)
2. 0,15 ml
de 10X MOPS
3. 0,24 ml
de formaldeído 37%
4. 0,1 ml
de H2O-DEPC autoclavada
5. 0,1 ml de glicerol (autoclavado)
Preparo da amostra para eletroforese:
1. Separe as alíquotas de RNA a serem submetidas a
eletroforese, de acordo com a quantificação feita previamente.
2. Se o volume por superior a 5 ml, reduza-o a 5ml no dessecador a vácuo. Se for inferior a 5 ml,
complete com água para 5 ml.
3. Adicione 25 ml de “loading buffer”
4. Aqueça a 65°C
por 15 min (para desnaturar) e ponha em seguida no gelo.
5. Se for corar com brometo de etídeo, acrescente à
amostra 1 ml de solução de brometo de etídeo (1mg/ml) em H2O-DEPC
autoclavada. Se for corar com SYBR Green, coloque a amostra nas poças do gel,
previamente coberto com tampão MOPS 1X (diluído em H2O-DEPC autoclavada), sem a
adição do brometo de etídeo.
6. Não é obrigatório o uso de marcador de peso
molecular para RNA, porque como se trata de RNA total, os RNAs ribossômicos,
muito abundantes, servem para indicar o tamanho das moléculas em cada posição
(rRNA 28S ~ 5kb; rRNA 18S ~ 2kb).
7. Corra a eletroforese até o corante percorrer
2/3 a
3/4 da extensão do gel.
8. Visulize no transiluminador UV e fotografe (utilize
uma régua transparente a UV durante a fotografia, para definir a posição das
moléculas de RNA), ou
9. Core com SYBER Green II, do mesmo modo descrito
para DNA. visualize no transiluminador ou faça o “scanning” no STORM.
Se desejar, transfira o RNA para
membrana de nylon (“northern-blot”).
O gel é útil para se verificar a
integridade do RNA (avaliada pelo aspecto do RNA ribossomal) que, na área de
diagnóstico, quase sempre será usado para transcrição reversa e PCR (RT-PCR).
Transferência para membrana (Southern-blot):
“Southern-blotting” é a
transferência de fragmentos de DNA do gel de eletroforese para uma membrana que
serve de suporte. A transferência e o tratamento subseqüente (secagem e
exposição a luz ultravioleta) resultam em imobilização de fragmentos de DNA na
membrana, de forma semi-permanente. Após a imobilização, o DNA pode ser
submetido a análise por hibridização, que permite a identificação de bandas
contendo moléculas de DNA similares à molécula de ácido nucleico utilizada como
sonda.
Os tipos de membrana mais comumente
utilizadas são as de nitrocelulose e as membranas de nylon, com ou sem cargas
positivas. As membranas de nylon são mais freqüentemente empregadas porque são
mais resistentes, permitindo o uso sucessivo de várias sondas. A membrana de nitrocelulose, no entanto, em
algumas situações pode fornecer menos “background” (ligação inespecífica da
sonda).
O método básico de transferência é a transferência por
capilaridade, usando um tampão com elevada concentração de sal, o que permite a
ligação do DNA à membrana. Na presença de elevada concentração de sal o DNA
fica preso à membrana durante a transferência, mas não de forma permanente. A
imobilização permanente (ou semi-permanente) é obtida pela exposição a luz UV
(membrana de nylon, com a qual o DNA estabelece ligações covalentes) ou secagem
prolongada (2 horas) em forno à vácuo a 80oC (membrana de nitrocelulose,
com a qual o DNA estabelece interação hidrofóbica relativamente fraca, não
covalente).
O
protocolo de transferência é dividido em três etapas:
1. O gel de agarose é tratado com
soluções que promovem a despurinação (necessária apenas quando se pretende
transferir eficientemente moléculas maiores que 5 kpb; esse procedimento remove
purinas introduzindo “nicks” nas fitas de DNA), desnaturação (separação das
fitas da dupla hélice, fundamental para que o DNA se ligue à membrana e à
sonda) e neutralização (quando o pH do gel cai a valores menores que 9, o que é
fundamental para que o DNA se ligue à membrana).
2. A segunda etapa é a transferência
propriamente dita.
3. Finalmente, o DNA é imobilizado na
membrana.
Despurinação: tratamento com HCl - 0,25M.
n
Deixe o gel mergulhado nessa
solução, sob agitação leve e contínua, por 30 minutos, à temperatura ambiente
(TA).
Desnaturação: tratamento com solução de
NaCl 1,5M/NaOH 0,5M
n
Deixe o gel mergulhado nessa
solução, sob agitação leve e contínua, por 20 minutos (TA)
Neutralização: tratamento com solução de NaCl 1,5M/Tris.Cl 0,5M, pH 7,0.
n
descarte a solução denaturante
e enxague o gel com água MiliQ. Em seguida deixe o gel mergulhado na solução de
neutralização por 20 minutos. Troque a solução e lave por mais 20 minutos.
A
transferência propriamente dita é feita promovendo o fluxo de um tampão (20X
SSC ou 10X SSC) através do gel, em direção à membrana, de modo que o fluxo
dessa solução arraste as moléculas de DNA que ficam depositadas na membrana, já
que não a podem atravessr (poros de 0,45 um). Esse fluxo é induzido
aplicando-se uma camada espessa de papel absorvente sobre a membrana que está
em contado com o gel (ver esquema da montagem), ou por aplicação de pressão
negativa que força a passagem do tampão através do gel e da membrana.
A
transferência por capilaridade demora de 15 a 16 horas e a transferência por
pressão negativa, bem menos demorada, demanda 1 hora. Concluída a transferência, a membrana é
removida após a marcação das posições correspondentes às poças do gel de
agarose (assim se poderá medir a distância de migração e comparar com a
fotografia do gel, que foi feita com este ao lado de uma régua), e lavada em
solução 2X SSC por 5 min, para remover o excesso de sal e vestígios de agarose.
Em seguida a membrana é colocada entre dois pedaços de papel de filtro
(3MM-Watmann) e submetida a secagem em forno, a 80oC, por 20
minutos, para fixação por UV, ou por 2 horas, se não se usar UV. Terminado esse
passo, a membrana está pronta para hibridização
com uma sonda específica, e pode
ser guardada por tempo indeterminado, em ambiente bem sêco.
Fixação por UV: Expõe-se a membrana a 70.000
microjoules/cm2 de radiação UV, 254 nm, por 2 a 3 minutos (UV crosslinker - Amersham).
Estratégias
de Hibridização:
Definições importantes:
DNA com cópia única: são as seqüências de DNA que
ocorrem uma só vez no genoma haplóide (na verdade, nessa definição podem estar
incluídas seqüências com um número muito pequeno de cópias).
Seqüências repetidas de DNA: seqüências
que ocorrem várias vezes.
Complexidade: descreve o total de
seqüências diferentes em uma amostra de ácidos nucléicos; isto é, seqüências
que não pareiam entre si nas condições experimentais utlizadas.
Quando se trata de RNA, complexidade tem o mesmo significado,
independente da abundância de cada transcrito.
Desnaturação: separação de seqüências
complementares de ácidos nucléicos.
Reassociação ou renaturação: o
restabelecimento de pontes de H+ entre duas seqüências complementares,
previamente separadas.
Estringência: se refere às condições físicas em
que a desnaturação ou a renaturação ocorre. Os fatores físicos que interferem
com a estringência serão discutidos adiante.
“Zippering”: descreve o fenômeno que
ocorre logo após o início da reassociação. Há formação de um núcleo de
reassociação seguida de rapidíssimo pareamento do restante da seqüência.
Hibridização: utilizado atualmente para descrever
o pareamento de bases (formação de duplexes) entre seqüências específicas, a
partir de qualquer combinação de fragmentos de ácidos nucléicos (DNA ou RNA),
usualmente “in vitro”.
“Melting Temperature” (Tm): é a
temperatura na qual as fitas de ácidos nucléicos que se pareiam entre si
estão desnaturadas em 50%. É um parâmetro útil para avaliação da
estabilidade da dupla-fita (seja DNA-DNA, DNA-RNA ou RNA-RNA).
Fatores que afetam a
velocidade de reassociação
1. Temperatura: a
velocidade máxima é alcançada a cerca de 25oC abaixo do Tm.
2. Concentração do sal: em concentração NaCl abaixo de 0,1 M, um
aumento de 2 vezes na concentração aumenta a velocidade de reassociação entre 5
e 10 vezes. A velocidade continua a subir até
concentração de sal de cerca de 1,2 M, que corresponde a cerca de 7
vezes a concentração numa situação padrão. Cátions divalentes, que estão frequentemente
presentes como impurezas nas soluções, têm um efeito muito mais intenso que os
cátions monovalentes, tornando necessário o uso de EDTA como quelante em
situações em que se deseja uma
“estringência” elevada.
3. Pareamentos imperfeitos (“mismatch”): para cada 10% de
pareamentos imperfeitos a velocidade de reassociação é reduzida de um fator de
aprox. 2 vezes, quando a reação ocorre em temperaturas 25oC abaixo
do Tm.
4 .Comprimento do fragmento:
4.1.
Reassociação de DNA-DNA: a velocidade de “zippering” é muito rápida quando
comparada à velocidade de formação do pareamento inicial (nucleação). Se os
fragmentos complementares são do mesmo tamanho, a velocidade de reassociação se
eleva com a raiz quadrada do comprimento (número de bases), relação válida para
fragmentos entre 100 e 100.000 pb. Isso aparentemente se relaciona com o maior
número de possibilidades de nucleação, contrabalançado pela maior restrição
espacial oferecida pelos fragmentos maiores. Quando os dois fragmentos são de
tamanhos diferentes, o efeito do comprimento sobre a velocidae de reassociação
depende de qual fragmento está presente em excesso. Se fragmento em excesso é o
curto, a velocidade de reassociação aumenta com o tamanho do fragmento escasso.
Se ocorre o contrário, há uma inesperada redução na velocidade de reassociação.
4.2. Reassociação
de RNA-DNA: na reação com excesso de DNA, a velocidade de reassociação é
cerca de 4 a 5 vezes menor do que a da
interação DNA-DNA.
5. Viscosidade: quando se considera o efeito da viscosidade,
deve-se distinguir entre viscosidade microscópica e macroscópica. Viscosidade
microscópica se refere ao micro ambiente em torno do DNA e é comumente alterada
pela adição de sacarose, glicerol ou perclorato de sódio. A macroviscosidade
depende da presença de polímeros (entre eles o próprio DNA). Aa viscosidade,
medida em viscosímetro, inclui ambas, a micro e a macroviscosidade. Na presença
de sacarose, glicerol, etilenoglicol e perclorato de sódio, a velocidade de
reassociação do DNA é inversamente proporcional à viscosidade. O aumento da
macroviscosidade (presença de ficoll ou dextran sulfato), por outro lado,
aumenta a velocidade de reassociação, porque provoca um aumento efetivo na
concentração de ácido nucleico em solução.
6. Composição de bases: praticamente não tem efeito sobre a velocidade
da reassociação.
7. Agentes desnaturantes: em solução aquosa, a velocidade ideal
de reassociação de ácidos nucléicos ocorre em temperaturas entre 60oC
e 75oC. No entanto, extensos períodos de incubação nessas temperaturas
podem levar a considerável quebra das moléculas de ácidos nucleicos, por
agitação térmica. Para reduzir a temperatura, mantendo a estringência, pode-se introduzir agentes desnaturantes,
como a formamida, que desestabilizam a dupla hélice, o que reduz a velocidade
de reassociação. Adição de 1% de formamida reduz o Tm em 0,72 oC e a
velocidade de reassociação em cerca de 1,1%. A redução na velocidade de
reassociação se deve a aumento da microviscosidade. A comparação da taxa de
degradação de DNA observada em solução aquosa, em temperatura elevada, com a
observada com formamida a baixa temperatura, mostra que a redução na degradação
compensa, em muito, a redução na velocidade de renaturação. Para híbridos
RNA-DNA a relação entre concentração de formamida e redução no Tm não é linear.
Na presença de formamida 50%, a estabilidade do híbrido RNA-DNA é maior que a
da dupla hélice de DNA. Essa é uma vantagem que pode ser explorada em técnicas
de microscopia, nas quais se pretende a
pareamento “in situ” com RNA mas não com DNA.
Fatores que interferem com a
estabilidade da dupla hélice
1. Pareamentos imperfeitos (mismatch): 1% de “mismatch” reduz a
estabilidade térmica em cerca de 1oC (avaliado pelo Tm).
2. Comprimento do fragmento: Quanto maior o fragmento, maior a
estabilidade da dupla hélice.
3. Concentração de sal: na faixa de concentração de 0,01-0,1M, o
Tm muda cerca de 16°C para cada 10 vezes de variação
na concentração de sal. O efeito é menor
em concentrações mais elevadas (1M). Os cátion divalentes têm um efeito muito
mais intenso que os monovalentes sobre o Tm.
4. Composição das bases: como o pareamento GC é mais estável (3
pontes de H) que o pareamento AT (2 pontes de H), o Tm de uma dupla-hélice está realcionado com
conteúdo de GC de acordo com a equação empírica:
Tm = 0,41 (%GC) + 69,3
O efeito da composição das bases, no entanto, pode ser eliminado na
presença de alguns solventes caotrópicos (por exemplo, o: cloreto de
tetrametilamônio 2,4 M).
5. “Criterion”: essa palavra descreve o limite mínimo de
fidelidade do pareamento de bases e comprimento da duplex, estabelecido pelas
condições de incubação. Na presença de tetrametilamônio, situação em que variações no Tm, decorrentes da composição
das bases, são eliminadas, o “criterion”
pode ser definido com precisão de 1oC.
A temperatura de incubação (Ti) para a hibridização ideal de
uma sonda pode ser calculada pela equação empírica:
Ti = Tm
- 15°C
Tm = 16,6
log[M] + 0,41[Pgc] + 81,5 - Pm - B/L - 0,65[Pf]
Onde:
M - é concentração molar de Na+,
até um máximo de 0,5M (1 X SSC contém 0,165M de Na+)
Pgc - é a porcentagem de
bases G e C na sonda
Pm - é a porcentagem de
“mismatched bases”
Pf - é a porcentagem de
formamida
B - é 675 para sondas de até 100
bases
L - é o comprimento da sonda em número
de bases
Obviamente, essa fórmula serve como uma orientação
incial, não substituindo o teste empirico, no laboratório.
Marcação de ácidos nucleicos a
serem utilizados como sondas:
Marcação com isótopo radioativo:
Escolha do isótopo:
32P : meia-vida: 14,3 dias
tipo de
decaimento: beta
energia: alta
33P: meia vida:
tipo de
deacimento: beta
energia:
média
35S: meia vida - ~75 dias
tipo de
decaimento: beta
energia:
baixa
125I: meia-vida - 60 dias
tipo
de decaimento: gama
energia:
média
3H: meia-vida: 12,35 anos
tipo de
decaimento: beta
energia:
baixa
Métodos de marcação:
1- “Nick-translation”:
Esse processo utiliza
DNAase I para gerar quebras (“nicks”) em cada fita da dupla fita de DNA. As ações de exonuclease 5’-> 3’ e de
polimerase 5’->3’da DNA polimerase I de E. coli são usadas para remover
trechos de DNA a partir das quebras e substituí-los por segmentos de DNA-fita
única, marcados pela incorporação de desoxirribonucleotídeos com átomos
radioativos. Pode-se utilizar qualquer desoxirribonucleotídeo marcado com 32P,
35S na posição alfa. Nucleotídoes marcados 125I, 3H
ou biotina também podem ser incorporados.
2-
Random oligonucleotide-primed labelling
3- Marcação com T4 DNA polimerase:
A T4 DNA polimerase
possui atividade de exonuclease 3’->5’ e atividade de polimerase 5’->3’.
Na ausência de fosfato de desoxirribonucleosídeos exógenos verifica-se apenas a
atividade de exonuclease que, em condições normais (temperatura e tampão
adequados), remove cerca de 25 nucletídeos/min. Quando os nucleotídeos são
adicionados, a atividade de polimerase predomina, e a extremidade previamente
removida é preenchida, agora com um ou mais desoxirribonucleotídeos marcados.
4- Marcação da extremidade com T4
polinucleotídeo quinase (T4 PNK):
A polinucleotídeo
quinase é usada para transferir o fosfato gama do ATP para o grupo 5’OH livre
na extremidade da molécula de DNA ou RNA. Essa enzima tem também uma atividade
fosfatásica. Duas reações são, portanto, possíveis: a)“forward reaction” em que
a enzima cataliza a fosforilação, após remoção do fosfato do terminal 5’ com
fosfatase alcalina, e b) “exchange reaction” em que a enzima cataliza a troca
de um fosfato 5’ previamente existente pelo fosfato gama do ATP. A reação de
troca tem que ser realizada na presença de excesso de ATP e ADP, para que se
consiga uma fosforilação adequada. Ambas as reações são mais eficientes quando
o DNA tem extremidade 5’ “protusa”. A extremidade 5’ “recuada” é fosforilada
com muito menor eficiência. Extremidades “rombas” (“blunt”) são marcadas com eficiência
intermediária.
O RNA é mais eficientemente marcado pela T4 PNK após
clivagem da base com NaOH.
5- Marcação da extremidade com
desoxinucleotidil transferase terminal (Terminal Deoxynucleotidyl Transferase -
TDT)
Essa enzima adiciona
desoxinucleotídeos na extremidade 3’ dos fragmentos de DNA. Tanto DNA-fita
dupla como DNA-fita única são substratos, na presença de cobalto como
co-fator. Até mesmo a extremidade 3’
“recuada” é substrato. Para evitar a formação de caudas de homopolímeros com
comprimento variável pode-se usar um
análogo do nucleotídeo que tenha o oxigênio do grupo 3’OH removido (alfa32P-cordycepin
trifosfato).
6- Marcação da extremidade com o
fragmento grande da DNA polimerase I de E. coli (Klenow fragment)
O fragmento Klenow da
DNA polimerase I de E. coli, que tem atividade de polimerase 5’-> 3’,
pode ser usada para preencher extremidades 3’ recuadas, que ocorram
naturalmente, ou que foram geradas por enzimas de restrição, tornando a
extremidade romba. Para isso se usa um ou
mais dos desoxirribonucleotídeos marcados a serem incorpoarados.
7- Uso de exonuclease III para
criar extremidades 5’ protusas (ou 3’ recuadas):
É uma alternativa à
utilização de T4 DNA polimerase.
Sistemas especiais de clonagem
para marcação de ácidos nucleicos com alta atividade específica:
1- Sonda universal com plasmídeo M13:
O DNA a ser utilizado como sonda deve
ser clonado nas séries pares dos vetores M13 (por ex.: M13mp8), pelas técnicas
usuais de clonagem em plasmídeo.. Um oligonucleotídeo de 13 mer (5’
GAAATTGTTATCC 3’), complementar a um segmento do plasmídeo, localizado na
região 5’ que flanqueia o sítio de múltiplos pontos de clonagem deste
(“multiple cloning site”), é usado como “primer” universal para síntese da fita
marcada complementar (fita (-)) à seqüencia da fita (+) do plasmídeo, feita
pelo fragmento Klenow da DNA polimerase I de E. coli. A síntese da fita minus
(-) não é completa, de tal modo que a inserção específica no sítio de clonagem
permanece como fita única, e, portanto, disponível para a hibridização com a
seqüência complementar que se deseja localizar.
2 - Sistema utilizando T3 e T7 RNA polimerase ou SP6 RNA polimerase
O fragmento de DNA a ser utilizado como sonda é
inserido no vetor plasmídeo SP6, derivado do pBR322, no qual foi inserido o
gene da RNA polimerase SP6 (fago de Samonella) e seu promotor. Algumas bases
adiante do promotor está o múltiplo sítio de clonagem, onde é inserido o DNA de
interesse.
Outra possibilidade, mais versátil que a anterior
devido à existências de dois promotores, é a inserção do segmento de interesse
no plasmídeo Blue Script, cujo múltiplo sítio de clonagem é flanqueado pelos
promotores das RNA polimerases T3 e T7.
Uma vez inserido o fragmento de DNA que servirá como
sonda, o plasmídeo é linearizado, utilizando uma enzima de restrição que corte
imediatamente após, ou bem na extremidade da seqüência específica clonada, e em
seguida porcede-se à síntese do RNA complementar, marcado com [alfa 32P]rNTP.
Esse sistema porque fornece uma grande quantidade de
transcrito marcado (até 10 mg para cada 1mg de
“template”), com elevada atividade específica.
O inconveniente é a instabilidade do RNA. A sonda deve ser tratada com
todos cuidados dispensados ao manuseio de moléculas de RNA. Não se presta
também a hibridização “in situ”, devido à facilidade de degradação.
Marcação não radioativa de
ácidos nucleicos:
Essas sondas eliminam
os cuidados relacionados ao uso de radioisótopos e têm a vantagem adicional de
serem estáveis, podendo ser estocadas por períodos de 6 meses a 2 anos.
Embora vários tipos de
marcadores não radioativos tenham sido descritos na literatura, a biotina
(GIBCO-BRL e outros) e digoxigenina (Boehringer Mannhein) são os mais comumente
utilizados e estão disponíveis comercialmente. Ambos podem ser facilmente
incorporados em sondas de DNA e ser detectados com os sistemas de detecção que
comentaremos em seguida.
Os desoxinucleotídeos marcados com biotina podem ser
detectados por complexos contendo avidina ou streptavidina, moléculas que
apresentam alta afinidade por biotina. A
avidina é uma glicoproteína (68.000 Da) derivada da clara do ovo, e a
streptavidina é uma proteína (60.000 Da) do Streptomyces
avidinii. A avidina ou a
straptavidina podem estar ligadas a uma
enzima, como a fosfatase alcalina, podendo esta ser detectada pela adição de um
substrato que resulte em um produto colorido, insolúvel. O sistema de detecção pode envolver,
alternativamente, a utilização de um anticorpo anti-biotina, anti-avidina,
anti-streptavidina, ou ainda anti-digoxigenina, seguida de um anticorpo
secundário conjugado a um fluorocromo ou a uma enzima.
Quando se usa anticorpo anti-biotina, o bloqueio da
membrana não pode ser feito com leite desnatado, porque este contém biotina e
causará intensos “background”.
DNA a ser marcado pode
ser um fragmento isolado, ou, como nas sondas para hibridização “in situ”, um
clone no fago, cosmídeo ou plasmídeo intacto.
Sondas Biotiniladas:
A incorporação de nucleotídeos biotinilados
(bio-11-UTP ou bio-16-UTP no lugar de dTTP e bio-14-dATP no lugar de dATP) é
feita através das técnicas previamente discutidas para marcação de ácidos
nucleicos com isótopos radioativos. Os métodos comumente empregados são
“nick-translation” ou síntese com oligonucleotídeos aleatórios biotinilados na
extremidade 5’(“random primed labelling”).
A Biotina é uma vitamina hidrossolúvel, que pode ser
covalentemente ligada à posição C5 do anel pirimidínico por meio de um “linker”
alilamina: bio-11-dUTP, ou bio-16-dUTP, no lugar de dTTP e bio-14-dATP no lugar
de dATP (os números referem-se ao número de grupos CH2 presente no
“linker”). O “linker” propicia uma
distância razoável entre a biotina e a base marcada, o que diminui a restrição
espacial decorrente da presença da biotina.
A
técnica de“Nick Translationresulta em incorporação”, em condições normais, de cerca de 50 nucleotídeos biotinilados por
cada 1000 pb.
Para “Random Primed Labelling” com oligonucleotídeos
não biotinilados normalmente se usam hexâmeros. Com oligonucleotídeos biotinilados
são usados octâmeros, pois a adição de mais dois nucleotídeos é necessária para
a estabilização do “annealing”, devido à instabilidade decorrente da restrição
espacial dada pela biotina na posição 5’terminal. Nesse último método, é comum adicionar-se à
reação de síntese pelo menos um desoxinucleotídeo biotinilado (bio-16-dUTP, por
ex.), para aumentar a eficiência da marcação. O DNA que serve de
“template” ( com 200 pb ou mais) tem que
estar linearizado e removido do vetor onde foi clonado, para maior eficiência.
Podem ser marcados de 25ng a 2 mg de DNA, mas a eficência da marcação
é maior com 25 a 50ng de DNA.
Digoxigenina-11-dUTP também pode ser incorporada ao
DNA tanto por “Nick Translation” como por “random oligonucleotide primed
synthesis”. Os protocolos são basicamente os mesmos utilizados para marcação
com biotina.
Essas sondas podem ser manuseadas normalmente,
evitando-se apenas extração com fenol e
desnaturação alcalina (a desnaturação deve ser feita pela incubação em
temperatura elevada, seguida de transferência imediata da sonda para o gelo).
Detecção por método
colorimétrico:
A biotina é detectada através de incubação com avidina
ou streptavidina, ou ainda com um anticorpo anti-biotina, quimicamente
acoplados a uma enzima que cataliza uma reação cujos produtos podem ser
quantificados por colorimetria (fosfatase, luciferase, peroxidase).
Um exemplo de sistema de detecção é a streptoavidina
conjugada a fosfatase alcalina, que se liga fortemente à biotina e é
visualizada quando se adiciona um substrato da fosfatase alcalina que produz um
precipitado colorido (NBT/BICP - nitroblue
tetrazolium/5-bromo-4-chloro-3-indoyl phosphate). O limite mínimo de detecção
com esse método colorimétrico é cerca de 2 pg de DNA.
Detecção por quimiluminescência:
O advento de técnicas
de quimiluminescência e de fluorescência tem tornado essas sondas quase tão
sensíveis quanto as marcadas com isótopos radioativos (métodos de detecção com
quimiluminescência são cerca de 5000 vezes mais sensíveis que os colorimétricos).
A fosfatase alcalina reage com um substrato
quimiluminescente que resulta na detecção altamente sensível do DNA alvo. O
substrato fluorescente é o DIOXETANE.
Este é clivado pela fosfatase alcalina o que resulta na emissão localizada de
luz que pode ser detectada por um filme de raio-X.
Os sistemas comerciais são vários, mas o princípio é o
mesmo em todos eles. O fragmento de DNA marcado com biotina, após hibridização
com a sequência alvo imobilizada em membrana de nylon, é ligado a fosfatase
alcalina biotinilada e, em seguida exposto, ao substrato de detecção.
Outra possibilidade é a utilização de sondas de ácidos
nucleicos marcadas com a própria fosfatase alcalina (ACES - GIBCO/BRL). Nesse caso, após a hibridização da sonda,
passa-se diretamente ao sistema de detecção (LUMI-PHOS PLUS - GIBCO/BRL).
A detecção com quimiluminescência deve ser feita em
membrana de nylon. Membranas de nitrocelulose não são indicadas, porque estas
promovem o esmaecimento do sinal luminoso (“quenching”).
Sondas marcadas com
substâncias fluorescentes - detecção por fuorescência e quimifluorescência:
Para síntese se sondas
fluorescentes, se utiliza um desoxinucleotídeo marcado com fluoresceína (ex.:
fluoresceína-11-dUTP - Fl-dUTP). O
sistema de marcação pode ser “Nick Translation” ou “Radom Primed Labelling”. O
sistema comercialmente disponível até o momento é por “Random Primed Labelling”
(Vistra Florescence - AMERSHAM). São
utilizados monômeros aleatórios como
“primers” para a reação de síntese da fita marcada pela Klenow DNA-polimerase.
A sonda sintetizada (com rendimento de cerca de 7ng/ml
por 25ng de DNA-template) pode ser estocada a -20oC por 6 meses ou
mais.
Após a hibridização, a
detecção pode ser feita imediatamente, sem amplificação do sinal, em
FluorImager (ex.: STORM - Molecular
Dynamics). Se for necessário, a membrana pode ser tratada com reagentes que
amplificam o sinal. O sistema de amplificação consiste de um anticorpo
anti-fluoresceína conjugado a fosfatase alcalina. Após a ligação do anticorpo,
adiciona-se o substrato da fosfatase alcalina (AttoPhos reagent - Amersham - fosfato orgânico aromático, cuja
estrutura é confidencial), podendo a membrana ser submetida a “scanning” em
FluorImager. Embora o sinal mais intenso
seja observado 24 horas após a adição do substrato, após 1 a detecção já é
possível.
Sites com diversos protocolos para diversas técnicas de biologia.
- PCR Primer - A Laboratory Manual
C.W.Dieffenbach
G.S.Dveksler
Editora: Cold Spring Harbor Laboratory Press – 1995
- Nucleic Acid Hybridization - A Pratical Approach
B.D.Hames & S.J. Higgins
Editora: IRL Press – 1991
- PCR - The Polymerase Chain Reaction
K.B. Mullis
F.Ferré
R. A. Gibbs
Revisado por James D. Watson
Editora: Birkhauser 1994
- PCR Applications – Protocols for Functional Genomic
Michael A. Innis
Davod H. Gelfand
John J. Sninsky
Academic Press – 1999.
- PCR – The Basics from Background to Bench
M.J. McPherson
S.G. Moller
Bios – Spring Verlag – 2000.
- PCR 2 – A Pratical Approach
M.J. McPherson
B.D. Hames
G.R. Taylor
PAS – A Pratical Approach Series
IRL Press – 1995.